Non-invasive fluorescence fundus imaging is an important tool for small animal in vivo retinal imaging in a wide array of translational vision applications, including the tracking of fluorescently tagged cells and blood vessels over time, as well as functional fluorescence imaging where calcium probes are used for monitoring retinal neuronal activity; these various applications require systems capable of imaging fine retinal structures in vivo. High resolution funduscopy solutions are primarily based on scanning laser ophthalmoscopes (SLO), with or without adaptive optics, or on wide-field imaging through low-NA objectives and topical endoscopes; two-photon in vivo imaging of the mouse retina was demonstrated only very recently using a very complex custom adaptive optics system.
In this study we use a model-based approach to analyze the requirements from an in vivo mouse retinal imaging system employing commercially available water-dipping objectives, and find that the mouse’s eye strongly constrains the range of imaging optics that can be used in this application. Our novel two-photon imaging solution is based on these design criteria, and uses widely accessible components to yield fundus images of optically sectioned, well-resolved fluorescent microstructures down to the cellular scale. Depth-scanning of the imaging plane is provided by an electronically tunable lens (ETL) integrated into the optical path with a fixed offset plano-concave lens, in order to avoid the perils of moving the objective. The system is found to provide major advantages in comparison to wide field imaging systems based on low NA objectives, and to allow long-term repeated imaging, in a simple, widely accessible design. Moreover, the system enables functional calcium imaging of repeated retinal responses to light stimulation using the genetically encoded indicator, GCaMP6s.
We constructed an intuitive simplified paraxial model for our optical system using published measurements of the mouse eye. Using this model we were able to analyze the position of the expected focal plane inside the eye when using multiple commercially available objectives (and their combinations with appropriate offset lenses). Following this analysis we chose to use long-focal length 10x objectives from Zeiss (0.45NA, WD=1.8 mm) and from Nikon (0.3NA, WD=3.5 mm) which were found to enable retinal imaging, with the latter providing a much wider working range leading to a superior ease of use. In order to obtain a model-based estimate of the lateral resolution in the retina, we developed a detailed ray tracing-based model that was implemented in the Zemax software, and calculated the lateral resolution of the focused beam on the retina to be ~1 µm.
To characterize the system’s axial scanning using this selected configuration, we repeatedly refocused the objective lens onto a single plane of vasculature in the living retina using a motorized z-stage for multiple ETL control values. These measurements provide a strong validation for the model’s predictions, allowing to translate changes in the optical parameters (objective position and ETL focal distance) to actual imaging depth.
Next, we acquired individual, optically sectioned planes of the retina through the mouse`s dilated pupil (Fig. 1A) by remotely scanning the retina with the ETL while keeping the objective static, and reconstructed a 3D image from these separate planes (Fig. 1B). Then, we tested whether the system can provide cellular-resolved images by intra-vitreally injecting an adeno-associated virus (AAV) expressing the genetically encoded calcium indicator GCaMP3. Transduced cells were clearly visualized in microscopic images, and by acquiring a subsequent fluorescence angiogram their location relative to the retinal vasculature could be determined and visualized (Fig. 1C, vasculature shown in false colors). By evaluating cross-sectional widths for the doughnut-shaped cell images (Fig. 1C inset, bottom), we estimated the system`s lateral imaging resolution to have an upper bound of ~3.5 µm. Therefore, we anticipate that the actual lateral imaging resolution will be somewhere in between this value and the predicted 1 µm value obtained from our optical model.
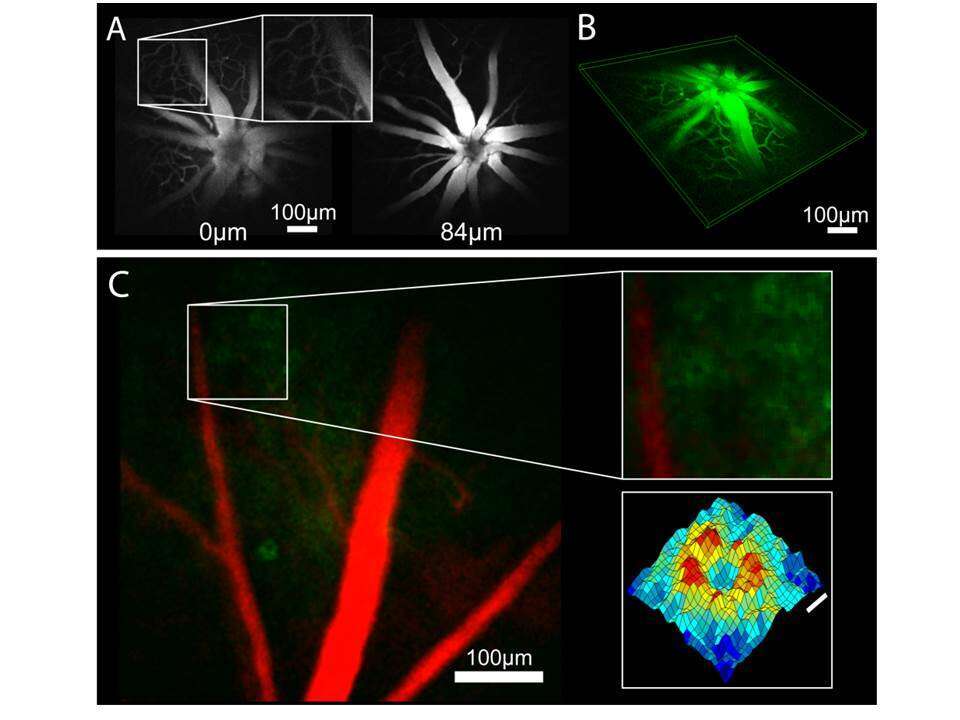
Figure 1. Imaging performance and characteristics. A) Fluorescent angiograms acquired with different ETL focal lengths (marked retinal depth labels are calculated from the model). B) 3D reconstruction of different retinal planes. C) False color image of retinal ganglion cells expressing GCaMP3 (green) and blood vessels (red, averages over 120 and 50 frames, respectively). Insets: Blowup of area with multiple cells (top) and surface plot of average cell image (bottom, scale bar=5 µm).
Finally, we examined the system’s ability to perform functional calcium imaging in vivo by recording the calcium dynamics from multiple retinal ganglion cells (RGCs) that express GCaMP6s in response to full-field flashes of light in the blue spectrum (Figure 2).
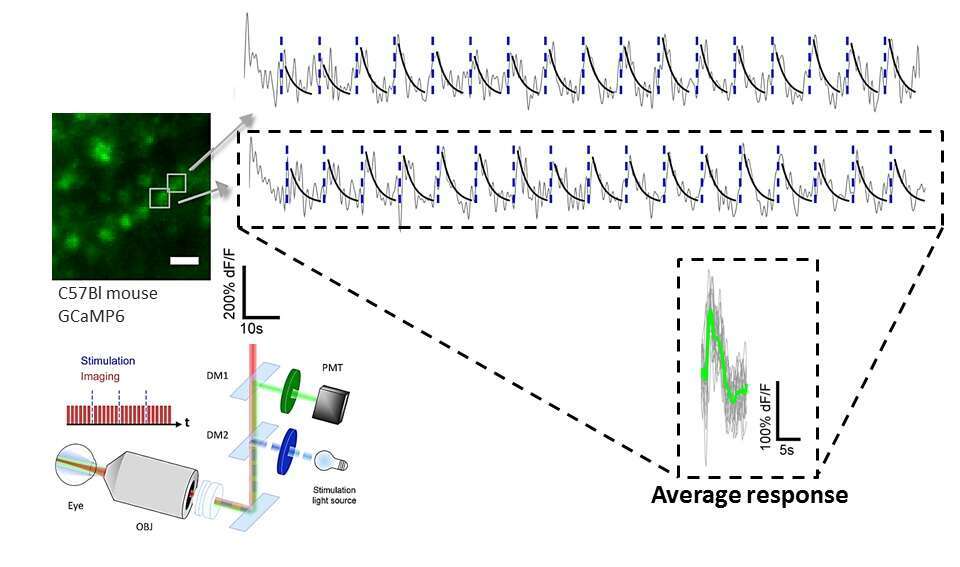
Figure 2. In vivo functional calcium imaging. Bottom: System schematic for the functional imaging experiments. Short pulses of blue light (10–20 ms) were flashed onto the retina as repeated scans of the FOV were acquired. Top: Representative calcium traces (gray) from two cells (marked in the average image on left) in response to the flashes of blue light (blue dashed lines) at 15-s intervals. Image scale bars = 20 µm.
In summary, we demonstrated the in vivo acquisition of optically sectioned two-photon laser scanning microscopic mouse fundus images using an intuitive optical model-based design, which can be easily integrated into essentially any existing microscope, and enable remote axial scanning, potentially up to multi-kHz rates. The system was found to provide exquisitely detailed 3D micro-angiograms and cellular-resolved images, whose axial location can be translated to “real-world” coordinates using the model. Furthermore, here we demonstrate for the first time the ability to carry out artifact-free two-photon functional imaging of neuronal retinal activity in the mouse in response to natural or artificial-optogenetic visual stimuli in the visible spectrum.
