
Synthesis and Evaluation of NPP1 Inhibitors as Potential Drugs for the Treatment of Osteoarthritis/CPPD Disease
2Department of Rheumatology, Tel Aviv Medical Center and the Faculty of Medicine, Tel Aviv
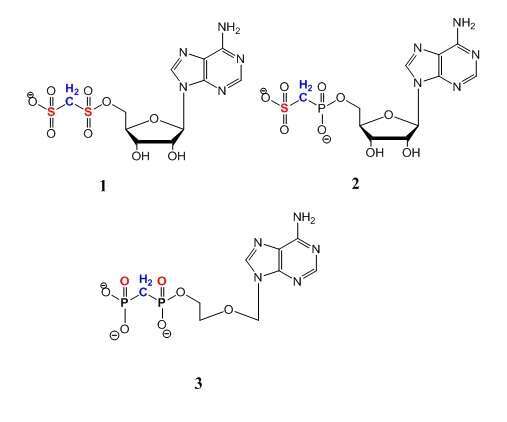
Overproduction of extracellular pyrophosphate due to hydrolysis of ATP by NPP1 leads to deposition of pathological Ca2P2O7·H2O (CPPD) in cartilage, resulting in a degenerative joint disease (CPPD disease) which has no cure. We explored the hypothesis that NPP1 inhibitors may be therapeutic agents for CPPD disease by inhibiting the hydrolysis of ATP. Specifically, we synthesized novel analogs of ADP (an NPP1 substrate) in which the Pa,b bridging oxygen atom is replaced by a methylene group, and a derivative where both Pa,b phosphate groups are replaced by sulfonate groups, 1. and the Pb phosphate is replaced by a sulfonate isoster, 2, In addition, we synthesized an ADP analog in which the ribose ring was cleaved, and the primary alcohol was substituted by bis-phosphonate, 3. To predict the ability of the derivatives to inhibit NPP1, we evaluated the affinity and selectivity of the analogs to Zn2+ involved in NPP1’s catalytic activity. We titrated the analogs with Zn2+- or Ca2+ ions and monitored the titrations by UV. Analogs 1 and 2 selectively coordinated Zn2+, and not Ca2+, and formed ZnL2 complexes, whereas analog 3 showed no affinity for Zn2+ and Ca2+ ions. Next, analogs 1, 2, and 3 have been evaluated for their inhibitory effect on NPP1 in human chondrocytes. Analog 3 proved to be a promising inhibitor reducing NPPase activity in human chondrocytes by 90% at 100 µM, vs. analogs 1, and 2 (40% and 30% inhibition at 100 µM). Analogs 1, 2, and 3, were found to be NPP selective showing no activity at TNAP. In summary, analog 3 was found to be an effective and selective NPP1 inhibitor in human chondrocytes. Since analog 3 is a poor Zn2+-chelator, we hypothesize that its inhibitory effect may be related to its high flexibility at NPP1’s catalytic site.

Powered by Eventact EMS