
INVESTIGATING THE SELF-ASSOCIATION PROCESS OF TEMPLATE MOLECULES USING A HIGH-LEVEL COMPUTATIONAL APPROACH, THE CASE OF CAFFEINE.
2Dipartimento di Fisica, Università degli Studi di Trieste
3Dipartimento di Scienze Chimiche e Farmaceutiche, Università degli Studi di Trieste
4Istituto Officina dei Materiali, Consiglio Nazionale delle Ricerche
Specific molecular recognition is one of the key properties of Molecular Imprinted Polymers (MIPs), and to achieve high selectivity the interaction between the template and the functional monomer(s) needs to be strong and stable during the polymerisation1. This interaction can be investigated using spectroscopic techniques but also computational methods. The latter have gained popularity in recent years, as a result of the exponential developments in computing powers and the advantages of saving resources and time. When evaluating the interactions it is also particularly important to also study the molecular structure of the units, and in particular their tendencies to form self-association complexes. (figure 1).
In this project we aimed to demonstrate the feasibility of using a computational approach to study the behaviour of a target analyte. We chose caffeine, a well known molecule found not only in coffee but also in many other products. Interestingly, it has been reported that in aqueous solutions caffeine can form aggregates, which would be expected to have an impact on any synthesised MIPs2. Therefore, it was decided to take this molecule as an example to applied an accurate computational methodology to study the self-association process3. Density Functional Theory (DFT) based methods were employed and different possible caffeine dimers were generated. Finally, the Gibbs free energy of association was calculated and compared with reported experimental values. To evaluate the applicability of the method for other compounds it was then decided to study the self-association process for paraxanthine, a structure similar compounds with caffeine but with different solubility properties. Finally, the computational data were used to find the suitable conditions for the synthesis in water of MIPs specific for caffeine, and the selectivity of the material was investigated.
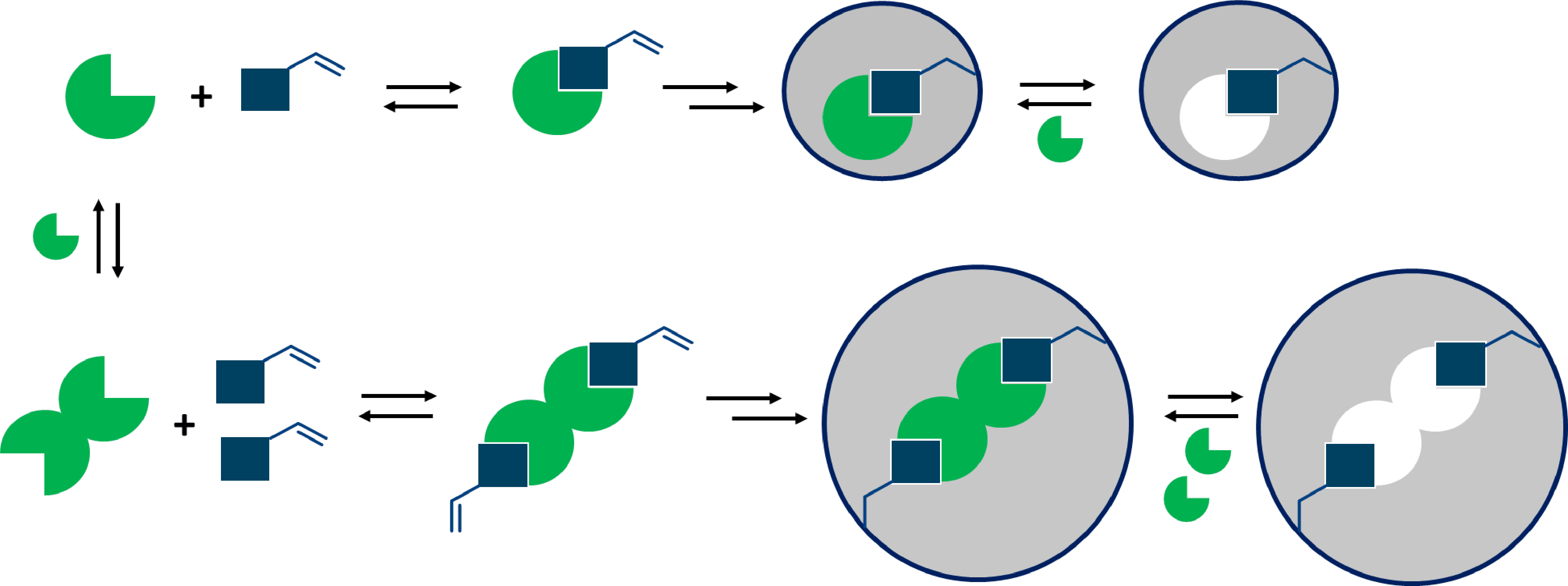
Figure 1. A schematic comparison between the synthesis of MIPs when the template is present as a free molecule, and when template aggregates are formed.
Reference
[1] Pellizzoni E., Tommasini M., Marangon E., Rizzolio F., Saito G., Benedetti F., Toffoli G., Resmini M., Berti F. (2016) Fluorescent molecularly imprinted nanogels for the detection of anticancer drugs in human plasma. Biosensensors and Bioelectronics 86:913–919.
[2] Tavagnacco L., Schnupf U., Mason P.E., Saboungi M. L., Cesaro A., Brady J.W. (2011) Molecular Dynamics Simulation Studies of Caffeine Aggregation in Aqueous Solution. The Journal of Physical ChemistryB 115:10957–10966.
[3] Di Tommaso D., Watson K.L. (2014) Density Functional Theory Study of the Oligomerization of Carboxylic Acids. The Journal of Physical ChemistryA 118:11098–11113.

Powered by Eventact EMS