
Copper-mediated deprotection of selenazolidine for cyclic peptides synthesis
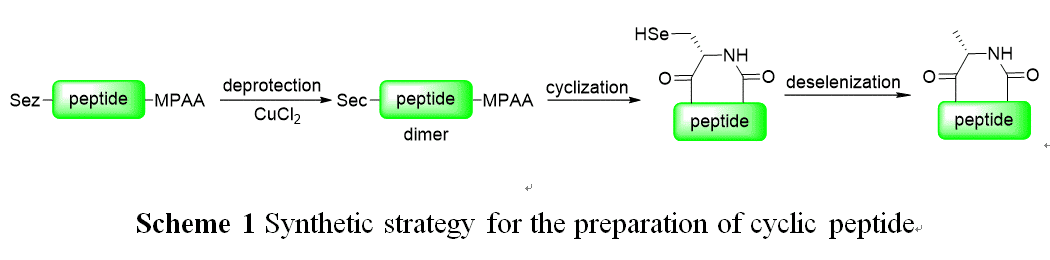
Cyclic peptides have high receptor-binding affinities and stability against proteolysis, due to their constrained conformation, hence they became promising synthetic targets for chemists.[1] The classical methods to prepare cyclic peptides are intramolecular amide formation with an activated acylation moiety.[2] However, the problems, including epimerization at C-terminal, polymerization and poorly-soluble protected peptides, limits their application for cyclization of large peptides. Intramolecular native chemical ligation of a unprotected peptide with thioester at C-terminal and Cys residue at N-terminal provide an efficient method for the synthesis of cyclic peptides.[3] Thiazolidine (Thz) and Selenazolidine (Sez), as protected forms of cysteine and selenocysteine, respectively, found wide application for synthesis of peptides and proteins. The common method for the deprotection of Thz and Sez is the use of MeONH2, which react with thioester and requires long period of reaction.[4] Herein, we developed an efficient method for the deprotection of Sez by metal ions, such as CuCl2, and applied it to the synthesis of cyclic peptide (Scheme 1).
References
[1] a) H. Kessler, Angewandte Chemie International Edition in English 1982, 21, 512-523; b) N. L. Daly, K. J. Rosengren, D. J. Craik, Advanced Drug Delivery Reviews 2009, 61, 918-930; c) R. Burman, S. Gunasekera, A. A. Strömstedt, U. Göransson, J. Nat. Prod. 2014, 77, 724-736; d) A. K. Yudin, Chemical Science 2015, 6, 30-49.
[2] a) J. S. McMurray, Tetrahedron Lett. 1991, 32, 7679-7682; b) J. Alsina, F. Rabanal, E. Giralt, F. Albericio, Tetrahedron Lett. 1994, 35, 9633-9636.
[3] L. Z. Yan, P. E. Dawson, J. Am. Chem. Soc. 2001, 123, 526-533.
[4] M. Jbara, S. K. Maity, M. Seenaiah, A. Brik, J. Am. Chem. Soc. 2016.
Powered by Eventact EMS