
Towards catalytic fluoroquinolones
2The Lorry I. Lokey Interdisciplinary Center for Life Sciences and Engineering, Technion – Israel Institute of Technology, Haifa, Israel
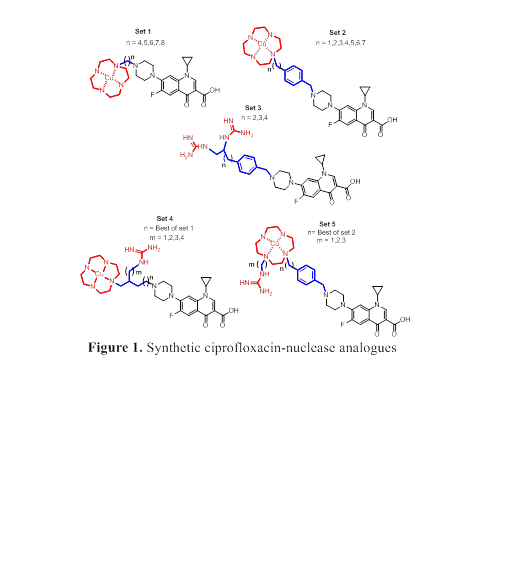
The appearance of bacterial strains resistant to multiple antibiotics has encouraged an extensive drive towards the goal of slowing down resistance development. One strategy is catalytic antibiotics, which seeks to mediate catalytic inactivation of the therapeutic target to form an inactive or dysfunctional entity. The possible benefits of this approach include: (i) increased potency, (ii) activity against resistant bacteria and (iii) reduced potential for the development of new resistance. Based on published structural and mechanistic data about the topoisomerase IIA-DNA complex (the target of fluoroquinolone antibiotics) and about artificial nuclease systems, we have rationally designed several ciprofloxacin-nuclease analogues with the potential to cleave phosphodiester bonds within the bacterial topoisomerase IIA-DNA complex, in such a manner as to cause bacterial chromosome fragmentation in a catalytic fashion (Fig. 1, set1-set5 structures). These ciprofloxacin-nuclease agents contain a Cu(II)-cyclen system, guanidine system or both of these systems, linked via a hydrophobic aliphatic or aromatic linker to the piperazine ring of ciprofloxacin, and are designed to catalyse DNA hydrolysis by leaving group activation, phosphate group stabilization, intramolecular-nucleophilic activation, or a combination thereof. The design, synthesis and preliminary biological evaluation of selected new designer structures will be presented.
Powered by Eventact EMS