
One more facet of DNA chemistry: Fast cationic isomerizations
In considering the chemistry of oxidative DNA damage, the deoxyribose-localized oxocarbenium ions C=O+ of DNA nucleotide units are accepted as the reactive cationic intermediates that initiate the reaction cascade leading to single strand breaks in this key biopolymer. However, this traditional consideration appears superficial – oxidized furanose fragments of syn-shaped nucleotide units are capable of rapidly isomerizing into much stable cyclonucleotide cations of another chemical functionality and another charge delocalization (e.g., amidinium cations for cytidine units or isouronic cations for thymidine units), as our studies show. An additional, nanosecond-scale isomerization is inherent to the 4ʹ-oxocarbenium ions of pyrimidine nucleotides: with disrupting the sugar ring, both syn and anti conformers rearrange into 1ʹ-azocarbenium ions C=N+ of these DNA units.
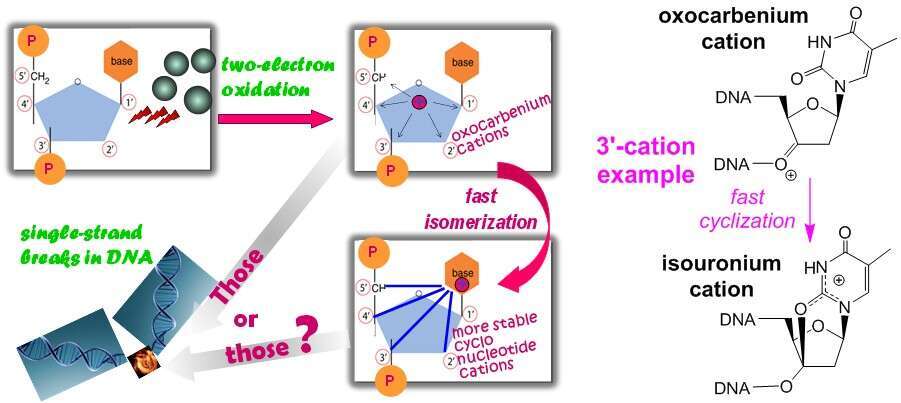
The revealed oxocarbenium ion chemistry predicts that chemical transformations of the 2e-oxidized sugar-phosphate backbone of DNA may be dissimilar in nucleic acids of different strand association [single-, double-, triple- and quadruple-stranded: ssDNA, dsDNA, tsDNA, and qsDNA, respectively]. The isomerization 4ʹ-oxocarbenium ion C=O+ ® 1ʹ-azocarbenium C=N+ ion can occur in any such a nucleic acid bearing the strand with a pyrimidine unit oxidized at the 4ʹ-position. Concerning the nucleotide cation-cyclizing isomerizations, the main structural factor, which prevents these cyclizations to unfold in dsDNA, is absent in ssDNA as well as Hoongsteen-pair sites of tsDNA and qsDNA. Clearly, such hydrolytically stable cationic sites may be impervious to DNA repair.
Powered by Eventact EMS