
Diastereoselective preparation of acyclic fragments bearing two stereocenters in a 1,5 relationship through an efficient three catalytic steps sequence
Despite substantial advances in the field of stereoselective organic synthesis, achievement of complete control over the stereochemistry in an acyclic system is still an ongoing challenge. The flexibility of a hydrocarbon chain and the diverse conformers that it can adopt represents a difficulty to discriminate one distinct configuration. The development of robust catalytic asymmetric methodologies for the creation of noncyclic stereocenters is therefore a highly desired goal.
In this work, we have envisioned a sequence that will allow the preparation of an acyclic fragment bearing two stereocenters in a 1,5 relationship. This motif is present in many natural diterpenoids such as α-tocopherol (vitamin e), vitamin k, crenarcheol and chlorophyll. Traditionally, these molecules have been prepared through iterative processes, installing one stereocenter at a time and requiring functional group interconversion between the iterations. Therefore, it seemed that a one-pot, diastereoselective and convergent reaction for the preparation of such a motif would provide a subsequent advance in their synthesis.
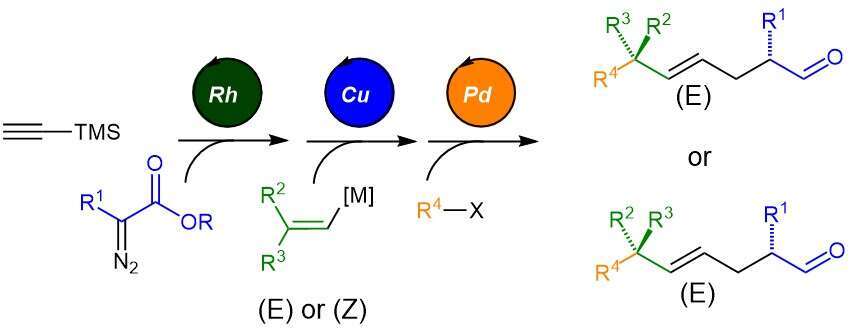
In order to achieve a diastereoselective route to the desired motif, we have devised a three catalytic step sequence. The two first steps allow the construction of a strained stereodefined cyclopropane which is then subsequently opened in the last step by a tandem Heck reaction to reveal the desired acyclic fragment. The sequence is designed in a convergent and modular way, combining four different building blocks that can be changed at will. Moreover, the relative stereochemistry in the product can be controlled by changing the stereochemistry of the alkenyl building block, rendering the reaction stereodivergent. This methodology was applied to the formal synthesis of α-tocopherol and epi- α-tocopherol.
Powered by Eventact EMS