
RyR2 Pathogenic Versus Benign Variants Can be Classifeid According to Thermodynamic Differences Using Normal Mode Analysis
2Faculty of Engineering, Holon Institute of Technology, Israel
Introduction: Cardiac Ryanodine Receptor (RyR2) is a calcium release channel in the sarcoplasmic reticulum of cardiac muscle cells. RyR2 plays a major role in the regulation of Ca2+ homeostasis. More than 1000 RyR2 misense mutations have been reported, out of which the vast majority remains of uncertain significance. It is known that proteins are dynamical objects, and Normal mode analysis (NMA) is a computational method for the assessment of protein flexibility based on its thermodynamic properties. Aims: to search for a novel method of variant classification by the effect on protein stability and flexibility.
Material and Methods: Pathogenic and likely pathogenic RyR2 missense variants were retrieved from ClinVar National Center for Biotechnology Information (NCBI) and benign variants from Exome Aggregation Consortium (EXAC). Each variant was created based on a solved cryo-EM structure of RyR2. NMA was performed for each one. Thermodynamic properties of the different variants were assessed by calculating a vibrational entropy. Next, a prediction algorithm was trained on the randomly chosen dataset of pathogenic and benign variants (~1010 combinations) and then tested on the remaining variants from the total dataset ((~1010 combinations).
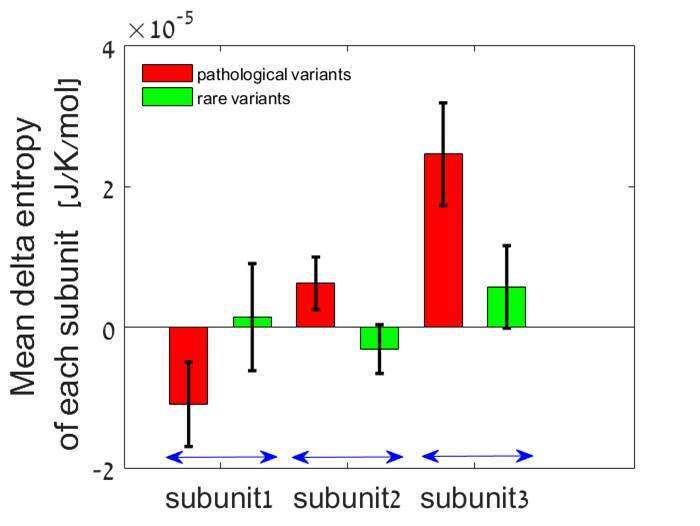
Results and discussion: 100 pathogenic/likely pathogenic and 73 benign variants were included in the analysis. Stability and the flexibility of RyR2 protein variants was calculated. The mean entropy was significantly higher for the pathological variants than the benign variants, and the number of RyR2 subunits, as well as number of residues in each subunit which were affected by a variant was significantly higher accordingly. The prediction algorithm was able to predict pathogenicity with a sensitivity of 0.88 ± 0.11 and specificity of 0.97 ± 0.10.
Conclusion: Pathogenic versus benign RyR2 variants significantly differ by mean entropy and number of affected residues and subunits. NMA can classify variants as pathogenic with high sensitivity and specificity.
Powered by Eventact EMS