Glycosylation
of intracellular proteins with O-linked N-acetylglucosamine (O-GlcNAc) is an
abundant and essential posttranslational modification in higher eukaryotes.
Inhibitors of O-GlcNAc transferase (OGT), the enzyme carrying out this
modification, are valuable tools to probe the cell biology of O-GlcNAc. Guided
by the recently reported X-ray structure of the ternary complex of human OGT (hOGT) with
substrate analogs [1], showing the
β-phosphate oxygen of UDP positioned at a distance of 3.4 Å from the C-β of the
O-GlcNAc-modified serine we devised a set of peptide−UDP conjugates as the
first generation of hOGT bisubstrate inhibitors.
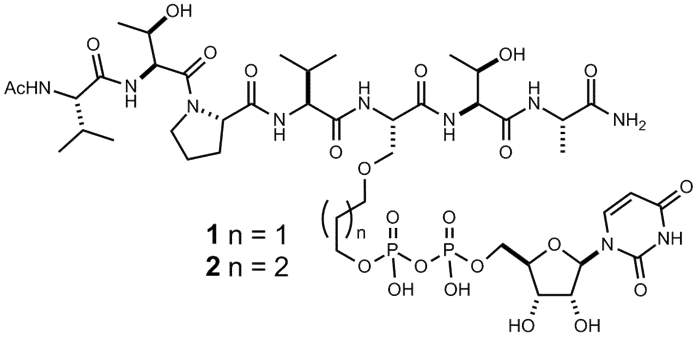
The
acceptor serine hydroxyl in the substrate heptapeptide VTPVSTA was linked
to the β-phosphate of UDP via 3 or 4 carbon atom tethers to retain the spatial arrangement seen in the
Michaelis complex, with
GlcNAc pyranoside ring being omitted from the inhibitor structure. Thus obtained
inhibitors 1 and 2 bind the enzyme with micromolar
affinity (Kd = 7.9 µM and 4.9
µM respectively) and inhibit glycosyltransfer onto a protein substrate, TAB1. A
co-crystal structure of hOGT and inhibitor 1 reveals the ordered linker with both peptide and UDP moieties occupying
positions equivalent to the ternary substrate complex. These findings prove the
validity of the bisubstrate inhibition concept for OGT and lay a foundation for
the development of the next generation of bisubstrate inhibitors.
[1] Schimpl, M.; Zheng, X.; Borodkin, V. S.; Blair, D. E.; Ferenbach, A. T.;
Schuettelkopf, A. W.; Navratilova, I.; Aristotelous, T.; Albarbarawi, O.;
Robinson, D. A.; Macnaughtan, M. A.; van Aalten, D. M. F. Nature Chemical Biology 2012,
8 (12), 969-974.
