1H solution saturation transfer difference spectroscopy (STD NMR) epitope maps experimentally rank hydrophobic binding interactions between substrate and enzyme under thermodynamic equilibrium. This approach is complementary to X-ray crystallographic structural techniques in that it can pull out and rank the hydrophobic interaction spots that matter in binding. In this study, one substrate is used to map and compare binding between a wild type xylanase XT6 and E159Q, a single hetero-atom mutation where -COOH (glutamic acid) is changed to -CONH2 (glutamine). The data show that primary binding across the -2 -1 subsites is preserved. However, a number of subtle changes are clearly discernible. The strongest STD NMR effects are seen between wild type XT6 and the H-5 hydrogen atoms at the -1 binding subsite. These hydrogen atoms fit into a pocket created by two of three highly conserved Trp residues. In the mutant, the most significant enzyme-substrate interactions are altered and weaker, the interactions are more similar (less of a “spread” is observed), and only the β-anomer binds.
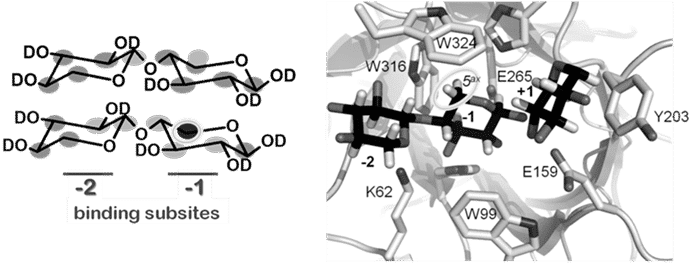
We hypothesize about the mechanistic effects of the mutation. In the superimposition of the apo and xylopentaose-XT6 complex structures (pdb coordinates 1R85 and 1R87, respectively) the side chains overlap. A notable exception is K62, occupying two degenerate positions in the apo structure. Only one configuration is maintained in the complex, with the ε-amino group of K62 pointing towards the middle of the xylose ring at the -2 subsite. In the acid/base catalytic mutant (E159Q) an important K62 – E195 electrostatic interaction is lost. Without this restraint, we propose a greater freedom of motion of the critical protein residues responsible for ensnaring xylose. This increased motional space allows for the same average chemical environment (chemical shift), but attenuates dipolar interactions underlying the saturation transfer measured by STD NMR, consistent with all the observed experimental findings.
