The reduced concentrations of the neurotransmitter acetylcholine (ACh) is a major feature of Alzheimer's disease (AD). Hence, acetylcholinesterase (AChE), which hydrolyses ACh, has been a main drug target for AD. Butyrylcholinesterase (BChE), another major cholinesterase (ChE), also hydrolyses ACh and its activity increases progressively in AD patients [1]. However, no therapy based on selective BChE inhibition is available. Moreover, abnormal expressions of BChE have been observed in various tumors [2]. These findings suggest that BChE may be used as a multifaceted therapeutic target for AD/cancer.
Our group previously showed the selective BChE nanomolar inhibition and low acute toxicity of novel 2-acetamidopurine nucleosides comprising a bicyclic sugar moiety [3]. Purine derivatives have also been reported as protein kinase inhibitors [4], namely cyclin-dependent kinases, which regulate cell cycle progression and play important roles in tumorigenicity.
These aspects prompted us to focus on the synthesis of new purine nucleosides for further bioactivity evaluation.
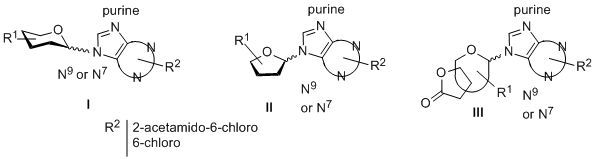
In this communication we present the synthesis of compounds of types I-III, in which a pyranose/furanose moiety or a bicyclic sugar lactone is linked to a purine via a C-N9- or C-N7-nucleosidic bond. Glycosyl donors including 1-O-acetyl or 1-thiophenyl derivatives, or methyl glycosides were used for the Lewis acid-promoted coupling to the silylated nucleobase. Variations on the substitution patterns of the sugar ring were made. Furanosyl nucleosides (II) were structurally chosen to target the ATP-binding pocket of protein kinases. The influence of the lactone ring on the stereochemical outcome of the N-glycosylation leading to III was investigated. Preliminary results of the biological activity will also be disclosed.
[1] Greig, N.H. et al. Int. Psychogeriatr. 2002, 14, 77-91
[2] Bernardi, C.C. et al. Cancer Genet. Cytogenet. 2010, 197, 158-165.
[3] Marcelo, F. et al. Bioorg. Med. Chem. 2009, 17, 5106.
[4] Veselý, J. et al. Eur. J. Biochem. 1994, 224, 771-786.